Single-Cell RNA Sequencing (scRNA-seq) has been instrumental in unraveling critical insights within mammalian systems, enriching our comprehension of transcriptional diversity across cell types and states. Equally pivotal is the need to uncover expression heterogeneity at the single-cell level in bacteria and microbiomes.
While scRNA-seq has made significant strides in elucidating the intricacies of eukaryotic cells, including plants and animals, and has become a cornerstone in biological research, microbial scRNA-seq technology faces formidable challenges. These obstacles include:
- Low mRNA Abundance: Bacterial mRNA content is strikingly meager, approximately two orders of magnitude lower than that found in mammals.
- PolyA Absence: Unlike eukaryotic counterparts, bacterial mRNA lacks the polyA structure at its 3' end, rendering its capture and analysis a complex endeavor.
- Short mRNA Half-Life: Bacterial mRNA has a substantially shorter half-life compared to mammalian mRNA, necessitating swift and efficient processing.
- Cell Wall Thickness: Bacterial cells boast thicker cell walls, posing obstacles to complete lysis and the efficient release of mRNA for analysis.
- Microbial Size Challenges: The diminutive size of microbial cells compounds the difficulty of isolating single cells for analysis.
In recent years, dedicated scientists have pursued technical innovations, resulting in the development of various bacterial single-cell RNA sequencing methodologies, including microSPLIT and BacDrop.
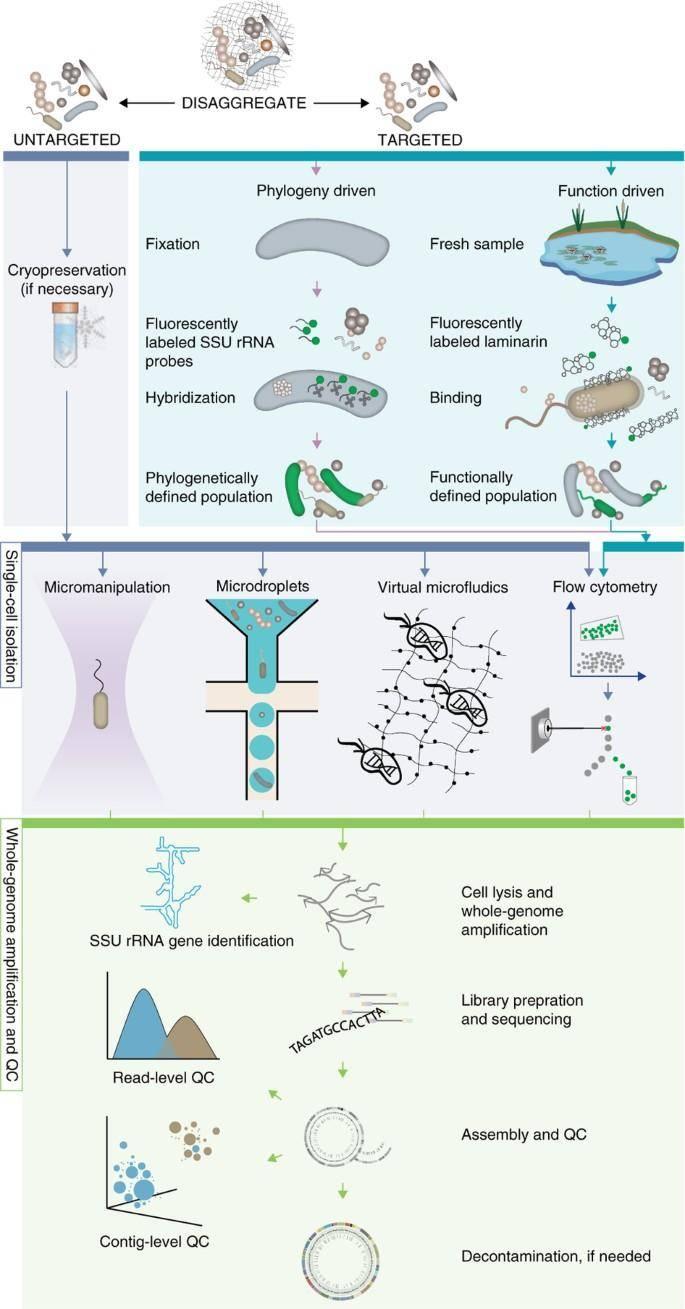
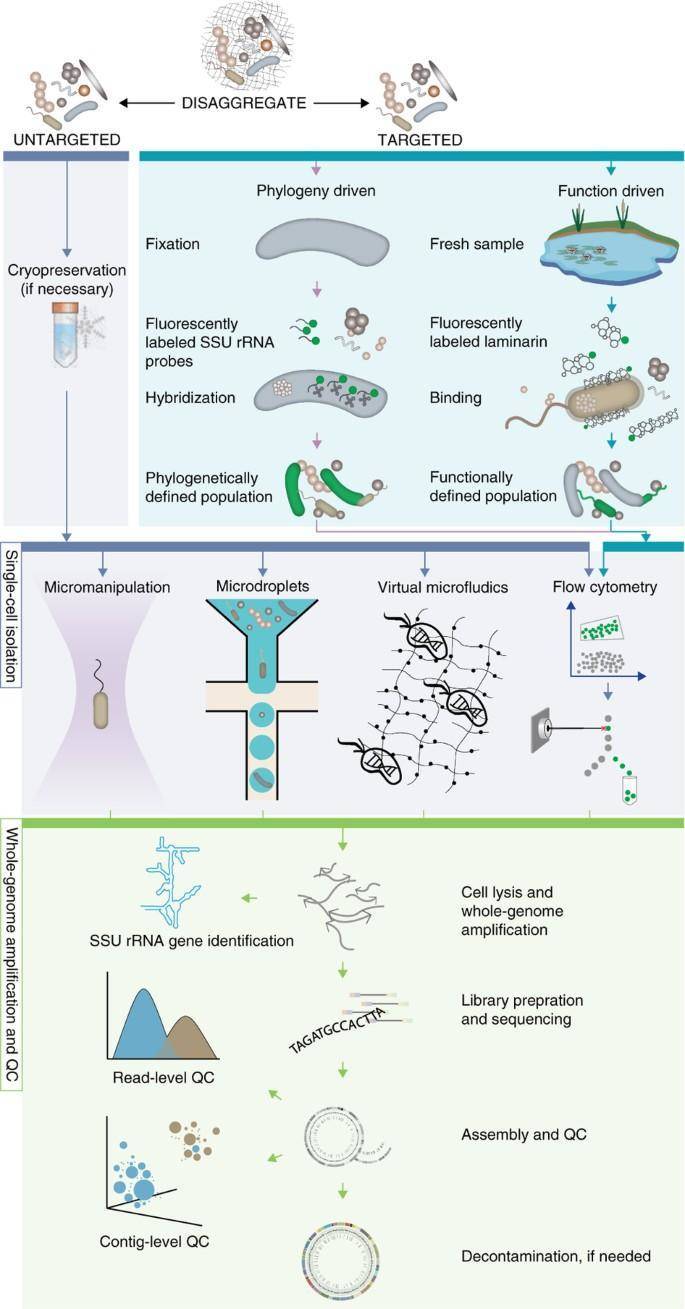 Single-cell sequencing and analysis workflow for standard (untargeted) and targeted single-cell sequencing approaches. (Woyke et al., 2017)
Single-cell sequencing and analysis workflow for standard (untargeted) and targeted single-cell sequencing approaches. (Woyke et al., 2017)
Eukaryotic Single-Cell RNA Sequencing (scRNA-seq)
In contemporary research, eukaryotic scRNA-seq has evolved into a widely employed technique, boasting a diverse array of experiments, analytical approaches, and several commercially available platforms.
Utilizing a macrophage infection model, scRNA-seq has unveiled the swift polarization of Salmonella-infected host cells, an observation that eluded mixed-pool RNA-seq techniques. This analysis further revealed that actively replicating Salmonella predominantly inhabits anti-inflammatory, susceptible M2 macrophages, while non-replicating bacteria are primarily localized in pro-inflammatory M1 macrophages. Notably, the rate of bacterial replication correlates with the extent of the host interferon response. Integration of scRNA-seq data with corresponding Dual RNA-seq data has established a link between the expression of a specific Salmonella virulence factor and macrophage polarization. Salmonella actively manipulates host cells to create a conducive environment for replication, mirroring the virulence strategy of Mycobacterium tuberculosis. These single-cell datasets represent a valuable resource for exploring the heterogeneity of different host cell types.
One limitation of conventional scRNA-seq is its dependence on poly(A) for capturing transcripts, resulting in the loss of information about transcripts lacking poly(A) tails, such as non-coding transcripts. To address this challenge, researchers have devised a method based on 3' end-joining junctions called Small-seq, enabling the detection of miRNAs, tRNAs, and snoRNAs in mammalian single cells. Another method, Holo-seq, examines both sRNAs and mRNAs within a single cell. While the current state of infection biology leans towards mRNA-focused investigations, researchers are increasingly turning their attention to interactions involving non-coding transcripts between microbes and their hosts. For instance, in Salmonella-infected human cells, lncRNA profiles exhibit more rapid changes than mRNAs, with certain host lncRNAs demonstrating the ability to reduce susceptibility to Salmonella infection. Moreover, some highly conserved bacterial sRNAs serve as critical regulators, while certain miRNAs contribute to host defense against pathogens, and others are actively exploited by Salmonella to facilitate pathogenesis. The study of non-coding transcripts in host-microbe interactions holds promise for further development in the future.
Bacterial Single-cell RNA Sequencing
Single-cell RNA sequencing (scRNA-seq) in bacteria has faced significant technical challenges that have impeded its widespread application in prokaryotes. These hurdles include factors such as small cell size, intricate cell separation, the presence of rigid cell walls, and the absence of polyA tails on mRNA molecules. As a result, the adoption of scRNA-seq in bacterial research is still in its infancy.
Several novel bacterial transcriptome detection technologies have been developed to address these challenges. Notable methods include MATQ-seq, MicroSPLiT, PETRI-seq, scDual-seq, and PatH-Cap technology. Notably, scDual-seq and PatH-Cap have been specifically designed for intracellular bacteria detection. For instance, scientists have employed MATQ-seq to analyze Salmonella typhimurium and Pseudomonas aeruginosa after isolating the cells. Comparative analysis with traditional mixed-pool RNA-seq data revealed that MATQ-seq excels in accurately capturing the expression patterns of growth-dependent genes.
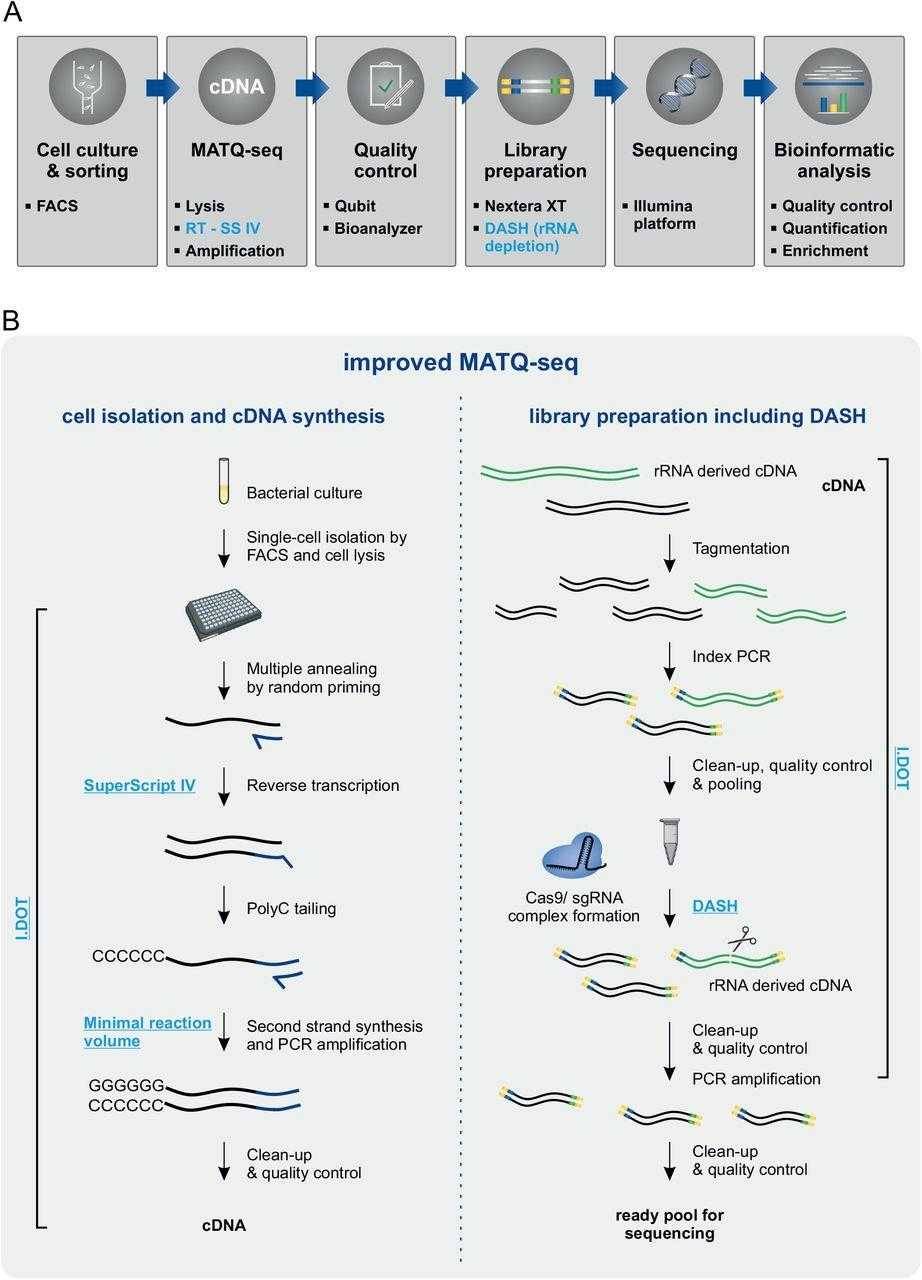 Improved MATQ-seq workflow for bacterial single-cell RNA-seq. (Homberger et al., 2023)
Improved MATQ-seq workflow for bacterial single-cell RNA-seq. (Homberger et al., 2023)
In another study, PETRI-seq was employed to investigate E. coli and Staphylococcus aureus, capturing approximately 200-300 mRNAs per bacterium. Meanwhile, microSPLiT was applied to individual Bacillus subtilis and E. coli cells, detecting an average of over 300 mRNA copies per bacterium. Although these methods achieved single-bacterial transcriptome sequencing, they still lag behind their eukaryotic counterparts in terms of cellular throughput, mRNA/gene capture rates, and sequencing depth of genes.
The field of microbial single-cell RNA-seq is rapidly evolving, with single-bacterial RNA-seq technologies for both Gram-negative and Gram-positive species still emerging. While the proof-of-concept stage is nearing completion, the next frontier involves studying natural microbial communities where individual bacteria may possess only a limited number of mRNAs, often not exceeding a few hundred. To advance this area, it is essential to develop efficient lysis and rRNA removal techniques for individual bacteria using CRISPR-based protocols. Additionally, the development of microfluidics-based devices to simplify sample handling is crucial.
Microbial communities can exhibit defined spatial organization with specific functions in both healthy and diseased states, necessitating the integration of scRNA-seq with advanced imaging techniques to decipher these relationships. Finally, unraveling the roles of microbial subgroups over time and in different spatial contexts will be pivotal for designing targeted therapeutic interventions.
References:
- Homberger, Christina, et al. "Improved bacterial single-cell RNA-seq through automated MATQ-seq and Cas9-based removal of rRNA reads." Mbio 14.2 (2023): e03557-22.
- Woyke, Tanja, Devin FR Doud, and Frederik Schulz. "The trajectory of microbial single-cell sequencing." Nature Methods 14.11 (2017): 1045-1054.